統計学から血液疾患のゲノム解析の道へ
がんゲノムの配列と構造の完全な再現を目指す(後編)
白石友一(国立がん研究センター研究所 ゲノム解析基盤開発分野 分野長)
2023.02.02
免疫回避の機序を解明し
再び『Nature』誌に

解析パイプラインの開発・メンテナンスを続けるだけでなく、統計学を専門とする研究者として、手法を開発するということも意識的に続けました。2013年には、コントロール検体を多数利用して後天的変異検出の感度を改善する方法論を経験ベイズ法という枠組みで定式化をした手法を実装・開発して論文にしました。この方法は、現在でもシークエンス解析パイプラインの根幹技術として使われています。また2014年4月から7カ月間、米国・シカゴ大学人類遺伝学科の客員研究員として勤務し、がんゲノム解析の統計手法に関する論文を1本仕上げました。テーマは少し専門的な説明になってしまいますが、mutation signatureという後天的塩基置換のパターンを明らかにする問題について、それまでに主流だったnonnegative matrix factorizationとは別のトピックモデルという方法論をベースに定式化を行なったものです。留学では、論文を1本仕上げるという目標を達成できたことも大きいですが、海外の研究の進め方を肌で感じて、自分の研究スタイルを見つめ直す良いきっかけになりました。メインで利用するプログラム言語をPerlからPythonに完全に切り替えたり、ソースコードのホスティングサーバーのGithubを積極的に使い始めるようになったことは、その時の経験によるものです。
2015年には、既に京大に移られていた小川先生がPD-L1に関する共同研究を始めました。このプロジェクトを中心になって進めた片岡圭亮先生は、既に成人T細胞白血病リンパ腫(ATL)においては免疫チェックポイント分子であるPD-L1の遺伝子異常による過剰発現を発見しており、このPD-L1の遺伝子異常が、がん細胞が免疫を回避するメカニズムの一端であることが推察されました。そこで、自分に任されたのはThe Cancer Genome Atlasの33種類のがん種1万例以上の検体について大規模な遺伝子解析をして、がん種横断的にPD-L1の構造異常を調査することでした。PD-L1はちょうど新しいがん治療として急速に注目を集めていた免疫チェックポイント阻害薬のターゲットの一つで、小川先生に「免疫チェックポイント阻害薬は本当にすごいんだ。(PD-1を発見した)本庶佑先生はノーベル賞を取るに違いない(実際に2018年に本庶先生はノーベル医学生理学賞を受賞されました)。とにかくこの研究はNatureを狙える」と発破をかけられたことを覚えています。1万例を超えるシークエンスデータを処理する解析パイプラインを開発して、PD-L1遺伝子異常の有無のスクリーニングを実施しました。その過程で、単純なゲノム構造異常だけではなく、HPVやEBVなどのウイルスがPD-L1遺伝子に挿入されることによる活性化もあるという追加の発見もしました。この研究結果は2016年の『Nature』誌に掲載されました。
クラウドを利用したゲノム医療の実現へ
すべてのがんのゲノム解析を究めたい
2018年4月に国立がん研究センター研究所の勤務となり、2020年4月からはゲノム解析基盤開発分野の分野長として、2つの大きなテーマを掲げて新たな仕事に取り組んでいます。

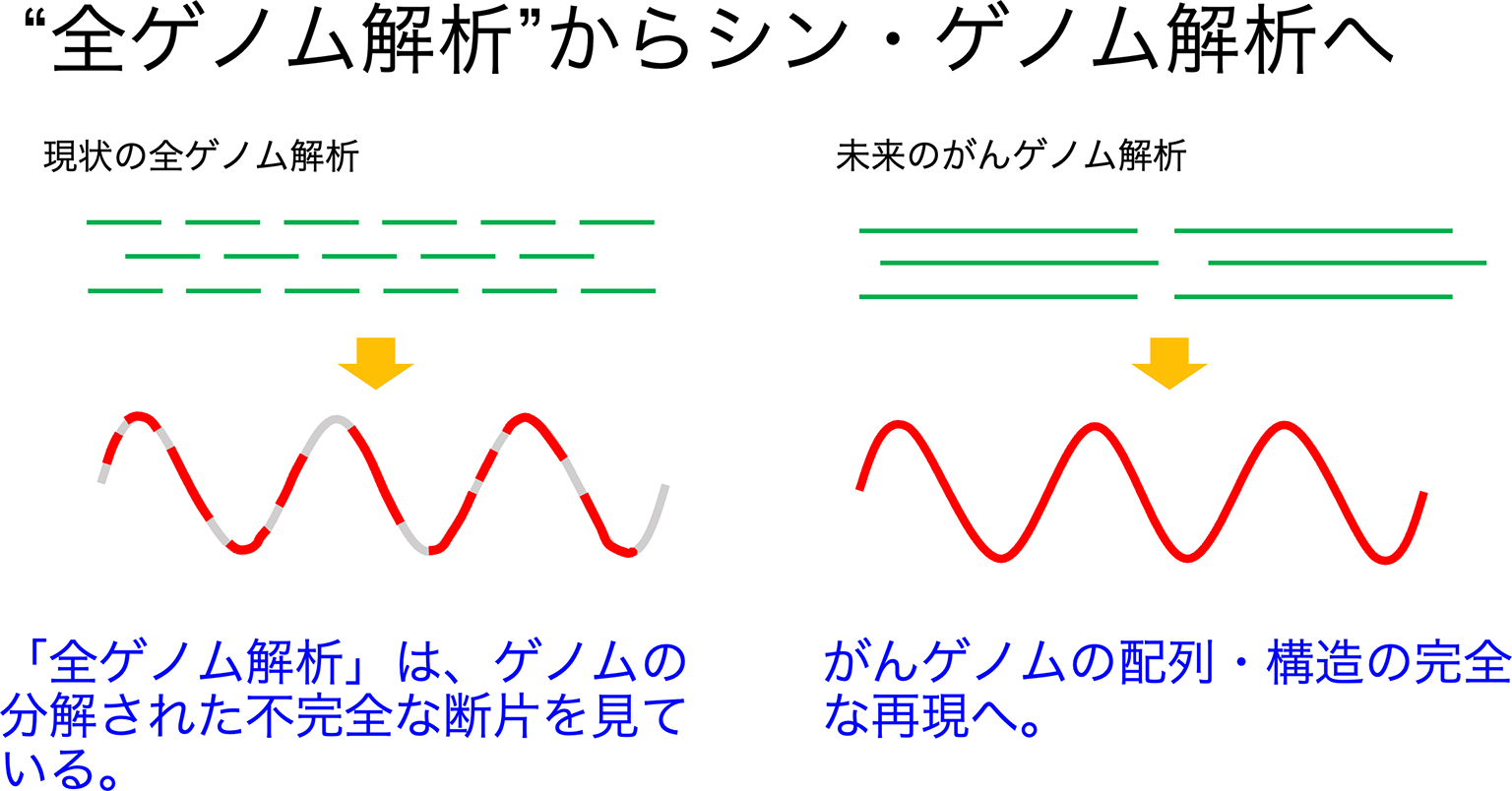
1つは、長鎖シークエンスデータを駆使して、がんゲノムを完全に再構成するプラットフォームを開発することです。現在進んでいる「全ゲノム解析」のプロジェクトのほとんどはショートリードのプラットフォームに基づいたもので、どうしてもゲノムのかなりの割合を占めるリピート配列の解析が困難になります。実際のところ、一般的な「全ゲノム解析」とされているものでは、ゲノム領域のかなりの部分はブラックボックスになっていて、ゲノムの分解された不完全な断片を見ているに過ぎず、本当の意味の「全ゲノム解析」からは程遠いものになっていると感じていて、そこに個人的には物足りなさをずっと感じていました。
最近の長鎖シークエンス技術の進展により、これまでは解明できていなかったセントロメアなどの高度リピート領域の配列を含めて、ヒトゲノムの完全配列が決定できるようになりつつあります。ただ、ロングリードといえども取得できる配列の長さは10kbpから最大でも1Mbpほどで、テロメアからテロメアまで安定的に繋がったゲノムを頑健に再構成する情報解析プログラムの開発が世界的に盛んに進められています。また、がんゲノムの場合は複雑な染色体間の転座、異数性、サブクローンの問題など、問題はさらに困難を極め、完全配列の再構成の試みはまだまだこれからだと思います。私が目指しているのは、色々な情報技術を駆使して、がんゲノムの配列を完全に再構成できる情報解析基盤(一般的な全ゲノム解析の限界を補完する真に完全なゲノム配列解析ということで、シン・ゲノム解析基盤と仮に呼びます)を開発することです(図1)。まずは、自分自身がその解析基盤のユーザーとなり、がんゲノムについての理解をさらに深めようと思いますが、これまでにGenomonでしてきたように開発したプラットフォームをがんゲノム解析のコミュニティが利用可能な形で提供して、研究・医療の進展に貢献できればと考えています。
2つ目は、国内・外で急速に蓄積が進むオミクスデータから、知識獲得を行なう解析基盤を開発することです。現在世界中で、研究・医療のためにゲノムシークエンス解析が盛んに行なわれています。まずは一次解析として、直接的な目的に応じた解析がなされるのですが、実際には生データが持つ情報量は膨大です。そこで、元データを世界中の研究者がアクセスできるレポジトリに登録して、再解析を促すということが一般的です。例として、前述のPD-L1のプロジェクトでも利用したThe Cancer Genome Atlasのプロジェクトでは、約10,000のがん検体についてゲノム、トランスクリプトーム、メチル化のデータは世界中の研究者が利用可能な形で提供され、これにより各国の様々な分野の研究者たちがありとあらゆる観点から再解析を進め、がん研究の大きな進展に繋がりました。今後もこうしたデータ共有・再活用の流れはますます進むはずです。その中で自分は、データの蓄積に合わせて自律的・自動的に「知識」が集積していくような未来を思い浮かべて、開発研究を進めています。ここで「知識」という曖昧な表現をしましたが、例えば何らかの疾患に関連する生殖細胞系列変異や、がんの進展に重要な働きを示す遺伝子異常と薬剤との関連などの情報を集積したデータベースなどを想像いただければと思います。そのための基盤開発の一環として、最近は大規模公共データからスプライシング変異をスクリーニングする情報解析基盤の開発をしています。
また、情報解析ができる人材の教育の必要性が議論されていますが、自分自身もできるだけ学生さんや研究員を受け入れて、このゲノム情報解析分野の発展に貢献できればと思います。最近はコロナ禍もあり色々と試行錯誤を続けていますが、情報解析においても単に数学やプログラミングができれば良いということではないと感じます。おそらく多様な問題に対する取り組み方(言語化することが難しい)の一つ一つが大切なのだと思います。最近読んだ本(「How to Get Rich」)で、“Specific knowledge can be taught through apprenticeships or self-taught.”とありましたが、結局のところ自分にできることは、一人一人と向き合い長い時間をかけて一緒に問題を解きながら、気づきを促すことだけかなと思うようになりました。
ゲノム解析をめぐる計測技術・情報技術の革新は目覚ましく、非常にエキサイティングな一方で、気を抜いているとすぐに遅れをとってしまうというプレッシャーも強くあります。PIとなりどうしても管理的な仕事も増えてしまいますが、その中でもできるだけ自分で手を動かすことは続けていこうと思います。
